Specific Research Focuses
Cystic kidney disease and fibrosis
Cystic kidney diseases comprise frequent (autosomal dominant polycystic kidney disease, or PKD) and rare forms (nephronophthisis, or NPHP) of cystic degeneration of the kidneys that invariably lead to complete loss of kidney function that requires dialysis or transplant. While cystic growth is the prevalent feature of PKD, nephronophthisis is characterized by intense inflammation and progressive fibrotic atrophy of the organ. In collaboration with Dr. Anton Jetten, we have shown that DNA damage and permanent cell cycle arrest of kidney tubular cells (replicative senescence) is the cause of the disease in our mouse model of NPHP. We are currently studying how replicative senescence induces inflammation and identifying possible molecular targets to slow down the progress of the disease.
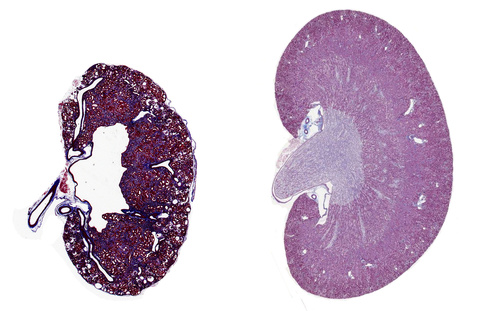
Kidney section of a 3 months Glis2 knockout mouse compared to a normal mouse of the same age. The size of the organ in the mouse in which the gene Glis2 was inactivated is decreased, due to the accumulation of collagen (blue stain) between the tubules and the atrophy of these structures, which result in diffuse cystic dilation and loss of kidney function.
Glomerular thrombotic microangiopathy in atypical hemolytic uremic syndrome
Thrombotic microangiopathy (TMA) is a condition characterized by damage of the small vasculature of multiple organs, platelet aggregation, and arteriolar and capillary thrombosis. The capillaries in the kidney glomeruli are invariably affected, which results in acute renal insufficiency that often progresses to complete loss of renal function. Most of the genetically-determined forms of TMA are secondary to defects in genes encoding factors that participate to the complement cascade and result in abnormal complement activation and endothelial damage. For the first time, we have identified mutations in the gene DGKE that cause TMA by mechanisms that are primarily unrelated to complement activation. We are currently using genetically modified mice and a wide array of biochemistry, cell and molecular biology techniques to understand the signaling pathways that are responsible for this disease, with particular focus on endothelial cells.
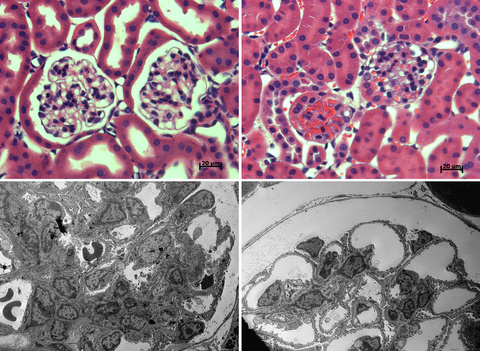
The upper pictures show how glomeruli of Dgke knockout mice (left) are more susceptible to develop microvascular occlusion after exposure to a sub-toxic dose of nephrotoxic antibodies, compared to normal mice (right). Transmission electron microscopy (lower panels) show glomerular capillaries are almost completely occluded in mice in which the gene Dgke was inactivated (left) compared to normal mice (right)
Understanding the mechanisms of kidney injury, repair and fibrosis
We investigated the state of the hedgehog pathway in the latter condition and showed that the hedgehog signaling is also activated in adult kidneys after injury. Further experiments that we conducted as a part of this project, showed the existence of a complex signaling between injured tubules and multiple interstitial cell populations that promote deposition of collagen in the interstitium, but also indirectly affect monocyte infiltration, epithelial cell survival, and organ repair.
Identifying genetic determinants affecting the risk of kidney stones disease
Kidneys stones are a diffuse problem in our society, affecting more than 10% of the adult population. Scientific evidence indicates that inheritance accounts for about half of the risk of common forms, but identifying the genetic contribution to the risk is not a simple task, given the contribution of environmental factors and the possibility that multiple genes are involved. In collaboration with Dr. Orson Moe and Charles & Jane Pak Center for Mineral Metabolism & Clinical Research, we are performing pilot rare variant association studies on extended cohorts of subjects affected with kidney stones.